Case Report
Potter Syndrome: A case study

Konstantinidou P1,2, Chatzifotiou E1,2, Nikolaou A1,2, Moumou G1,2, Karakasi MV2, Pavlidis P2 and Anestakis D1*
1Department of Histopathology, Laboratory of Forensic and Toxicology, Aristotle University of Thessaloniki, Greece
2Laboratory of Forensic Sciences, Democritus University of Thrace, School, Greece
*Address for Correspondence: Anestakis D, Department of Histopathology, Laboratory of Forensic and Toxicology, Aristotle University of Thessaloniki, Greece, Email: [email protected]
Dates: Submitted: 09 August 2017; Approved: 30 August 2017; Published: 31 August 2017
How to cite this article: Konstantinidou P, Chatzifotiou E, Nikolaou A, Moumou G, Karakasi MV, et al. Potter Syndrome: A case study. J Forensic Sci Res. 2017; 1: 063-067.
DOI: 10.29328/journal.jfsr.1001007
Copyright License: © 2017 Konstantinidou P, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Potter syndrome; Polycystic kidney disease; Oligohydramnios sequence
Abstract
Potter syndrome (PS) is a term used to describe a typical physical appearance, which is the result of dramatically decreased amniotic fluid volume secondary to renal diseases such as bilateral renal agenesis (BRA). Other causes are abstraction of the urinary tract, autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD) and renal hypoplasia. In 1946, Dr Edith Potter characterized this prenatal renal failure/renal agenesis and the resulting physical characteristics of the fetus/infant that result from oligohydramnios as well as the complete absence of amniotic fluid (anhydramnios). Oligohydramnios and anhydramnios can also be due to the result of leakage of amniotic fluid from rupturing of the amniotic membranes. The case reported below, concerns of stillborn boy with potter syndrome.
Introduction
Potter Syndrome is not technically a ‘syndrome’ as it does not collectively present with the same telltale characteristics and symptoms in every case. It is technically a ‘sequence,’ or chain of events - that may have different beginnings, but ends with the same conclusion. Below are the different ways that Potter Syndrome (A.K.A Potter Sequence) can begin due to various causes of renal failure. They have been given numbers to differentiate the different forms, but this system has not caught on in the medical and scientific communities [1].
Potter syndrome type I
This condition is inherited in an autosomal recessive character and occurs in one in 30 thousand births. The responsible gene detected on chromosome 6 and prenatal diagnosis in high risk groups is possible from the first quarter with trophoblast biopsy. Ultrasound, both kidneys appear swollen and echogenic with numerous cysts in the cortex, diameter <2mm. The strong echogenic attributed to the strengthening of ultrasound by tiny cysts. The disease is bilateral and symmetrical, but ultrasound imaging of the disease may not be possible until 24 weeks, and thus, continous scans are required to exclude the diagnosis. Other diagnostic criteria are the absence of bladder and oligohydramnios. The polycystic kidney disease falls into embryonic form (the most frequent), neonatal, child and adolescent, depending on the display time. The embryonic form results into death due to pulmonary hypoplasia and renal failure. Neonatal type has a poor prognosis with kidney failure in the first year of life. Childhood and adolescence lead to chronic renal failure, liver fibrosis and portal hypertension, while a kidney transplant is often required [2,3].
Potter syndrome type II
This form occurs in one in 1,000 births, while half the cases are associated with chromosomal abnormalities, genetic syndromes and other defects (mainly cardiac). By ultrasound examination, kidneys are displayed with multiple cysts of varying size, which do not communicate and echogenic layer. The diameter of the cysts determines the final size of the kidneys, which can be large, as in Formula IIa, or small, as in Formula IIb [4]. It can be bilateral, unilateral or relate to a portion of the kidney. In bilateral disease there is coexistence of anydramnio-oligohydramnios, whilethe bladder is absent. When it comes to bilateral disease, the prognosis is fatal, while in unilateral disease, the prognosis is good (although other urogenital anomalies may coexist at a large percentage). There is a wide range of views on the preventive kidney excision, while the vast majority of urologists adopt a conservative approach, as the affected kidney regresses and may disappear [5,6].
Potter syndrome type III
The disease is inherited in an autosomal dominant way and remains asymptomatic until the third to fifth decade of life. Unlike embryonic type, it is rarely diagnosed prenatally. Prenatal diagnosis refers to a few cases relating to fetuses with family history and forms that were fatal during neonatal and school age. One in 1,000 individuals carry the gene mutation on chromosome 16. Prenatal diagnosis is possible by trophoblast biopsy in the first quarter, while parents should be noted that the normal display of prenatal kidney does not exclude the manifestation of the disease during adulthood, characterized by highly enlarged kidneys, with numerous cysts of varying size. The amniotic fluid may be normal or decreased. In one third of cases cysts are observed in the liver, pancreas and spleen, while in one fifth of cases there is coexistence of intracranial aneurysms. The prognosis varies from neonatal death, hypertension (50-70%), Berry aneurysms (10-30%), to normal asymptomatic life [7,8].
Materials and Methods
The present incident described below involves a stillborn boy with the medical record of Potter syndrome, whose death was attributed to lesions (particularly in the lungs, liver, spleen and kidneys as the referral described) that may be related to the background history of Potter syndrome. The following biological samples were received in plastic containers with formalin by the authors:
• Tongue tissue sample
• Heart
• Lung tissue sample
• Liver tissue sample
• Spleen tissue sample/p>
• Intestine tissue sample
• Stomach tissue sample
• Kidneys
• Thymus
The tissue sections were stained with Hematoxylin and Eosin (H & E) staining technique. The procedure was:
• Deparaffinize the section: flame the slide on burner and place in the xylene. Repeat the treatment.
• Hydration: Hydrate the tissue section by passing through decreasing concentration of alcohol baths and water. (100%, 90%, 80%, 70%)
• Stain in hematoxylin for 4 minutes.
• Wash in running tap water until sections “blue” for 5 minutes or less.
• Differentiate in 1% acid alcohol (1% HCl in 70% alcohol) for 1 second.
• Wash in running tap water.
• Stain in Eosin for 2 minutes.
• Dehydrate in increasing concentration of alcohols and clear in xylene
• Mount in mounting media
After the staining procedure, the sections were examined under microscope and results were obtained [9].
Results
At relatively hypoplasmic lungs, extensive infiltrates were macroscopically observed with extensive bleeding, mild interstitial edema, cells with sparse eosinophilic material and areas of coincidence of the wall. Extensive presence of fibrous connective tissue was identified with dilated bronchioles which are locally outlined by eosinophilic material in the presence of the fibrin network, red cells, phagocytes, haemosiderin granules and clots within several bronchioles (Figures 1,2). Inflammatory infiltrates were also found, as well as vessels with thickened wall and red thrombi in the lumen of several of them.
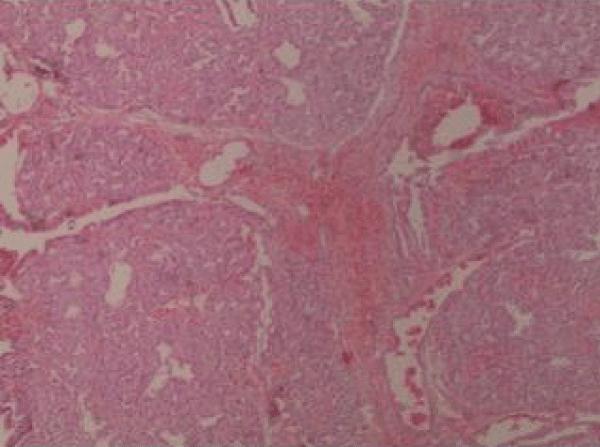
Figure 1: Lung tissue sample.
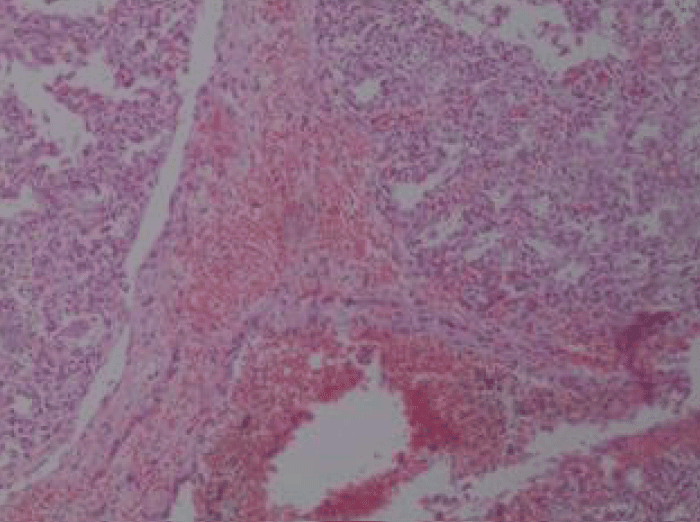
Figure 2: Lung tissue sample.
Extensive hemorrhagic infiltration, fibrotic and necrotic foci, inflammatory infiltrates and hemosiderin granules were also present at several positions in the kidneys (Figure 3). Regions with elongated, cumulative tubes were also detected with local presence of cysts. At a position a cavity was observed, in which a renal parenchyma section was recognized, which was connected via a thin ‘stem’ with the remaining parenchyma, as well as blood vessels with thickened walls and the presence of red thrombi.
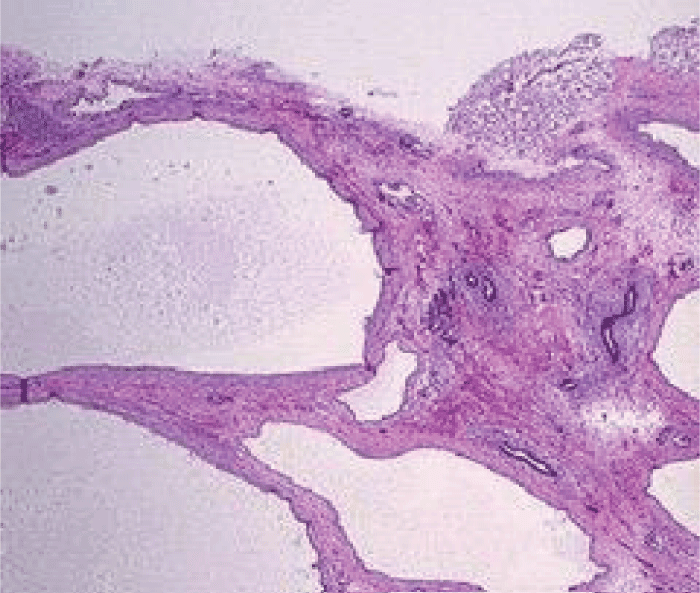
Figure 3: Polycystic kidneys
Discussion
As reported above, Potter syndrome varies in its types, symptoms and severity. In cases of stillbirth due to oligohydramnios, no treatment is available to date-just the diagnosis through amniocentesis, ultrasound or another form of prenatal control.
Patients who survive birth, however, can follow a course of therapies to manage the disease. These include nutrition control in newborns, through sufficient nasogastric feeding as well as management of electrolyte imbalances with specific medication. Also, regulation of anemia via oral or parenteral iron erythropoietin stimulating agents is required. Individuals may present with hypertension due to the activation of the renin-angiotensin system, handled by antihypertensive medicine which includes diuretics, beta-blockers, calcium channel blockers, and ACE inhibitors. Children with low glomerular filtration rate (GFR) are also provided with growth hormone, in order to grow in a healthier rate [10,11].
Despite the severity of the symptoms, the majority of the patients are in need of mechanical ventilation and chest tube placement for breathing support and for the treatment of spontaneous pneumothorax, due to pulmonary hypoplasia. In several cases, surgical care takes place such as the placement of a peritoneal dialysis catheter or a central venous line, vesicostomy in patients with posterior urethral valves or valve ablation, or even nephrectomy when a large size polycystic kidney is present. When the nutrition is inadequate, G-tube is placed [12].
References
- Boucher C, Sandford R. Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2). Eur J Hum Genet. 2004; 12: 347-354. Ref.: https://goo.gl/bpYdBp
- Klaassen I, Neuhaus TJ, Mueller-Wiefel DE, Kemper MJ. Antenatal oligohydramnios of renal origin: long-term outcome. Nephrol Dial Transplant. 2007; 22: 432-439. Ref.: https://goo.gl/5JiLmH
- Manoj MG, Kakkar S. Potter’s Syndrome-a fatal constellation of anomalies. Indian J Med Res. 2014; 139: 648-649. Ref.: https://goo.gl/KmdXXU
- Eccles MR, Stayner CA. Polycystic kidney disease-where gene dosage counts. F1000Prime Rep. 2014; 6: 24. Ref.: https://goo.gl/5FuqbP
- Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002; 13: 2384-2398. Ref.: https://goo.gl/55tYuA
- Lina F, Satlinb LM. Polycystic kidney disease: the cilium as a common pathway in cystogenesis. Curr Opin Pediatr. 2004; 16: 171-176. Ref.: https://goo.gl/KqGUS6
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006; 85: 1-21. Ref.: https://goo.gl/TB3fzT
- Tahvanainen E, Tahvanainen P, Kääriäinen H, Höckerstedt K. Polycystic liver and kidney diseases. Ann Med. 2005; 37: 546-555. Ref.: https://goo.gl/FnyLH9
- Kiernan JA. Histological and Histochemical Methods: Theory and Practice. 4th ed. Bloxham, UK: Scion. 2008.
- Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014; 75: 148-157. Ref.: https://goo.gl/1DcSJf
- Dias T, Sairam S, Kumarasiri S. Ultrasound diagnosis of fetal renal abnormalities. Best Pract Res Clin Gynaecol. 2014; 28: 403-415. Ref.: https://goo.gl/oEMkNT
- Horie S. ADPKD: molecular characterization and quest for treatment. Clin Exp Nephrol. 2005; 9: 282-291. Ref.: https://goo.gl/sfk5hQ